
Energy Lab 综述: 解析快充工况下的锂嵌入/析出的机理(1/2 锂嵌入机理)
“市场对便携式电子设备和电动汽车需求的不断增长,推动了锂离子电池朝着更高能量密度、更高安全性、更快的充电速度方向发展。然而,目前的高比能LIBs(锂离子电池)无法一直安全、高效地维持超高功率的能量输入,这主要是受石墨电极上的析锂的影响。本综述的目的是从底层原理和检测方法上去解析析锂诱因和实现对析锂的监测,使石墨负极能够支持更高功率的快充,同时提高LIBs的安全性。因此,本文将深入讨论石墨与Li+之间的相互作用,包括固体电解质界面膜(SEI)形成、Li+嵌层/析出行为。此外,探究Li+嵌层/析出动力学的认识历程以及在3种极端条件(高荷电状态、高充电倍率和低温)下的析锂机制也是本文全面研究LIBs析锂问题的重点环节。同时,本文总结了LIBs析锂引起的问题、析锂的检测方法,讨论了相关的知识空白,也为LIBs中析锂的后续研究方向提供了依据。”该综述发表在《Energy Lab》创刊号,文章的第一作者来自北京理工大学博士生杨毅,通讯作者为北京理工大学闫崇副教授和清华大学张强教授。

电池的发明使人类掌握了储存和释放电荷的能力。回顾一下商业电池发展的历程:铅酸电池由Planté于1859年发明,并由Faure于1881年进一步优化。铅酸电池由铅、氧化铅和硫酸组成,其电压可达2 V左右。虽然铅酸电池已经发明了160多年,但由于其低成本和回收的稳定性,铅酸电池仍然在电池消费市场占有压倒性的份额。镍镉电池是继铅酸电池之后应用最广泛的电池,无论在高温环境下还是在低温下,镍镉电池都比铅酸电池具有更好的循环性能。但由于镍镉电池成本高、存在记忆效应以及环境污染等问题,逐渐被后来开发的镍氢电池所取代。随后,新的电池体系不断被开发,逐步向便携、低成本、高能量密度、高安全性等方向发展。作为故事的主角,锂电池正一步步向前走来。从它的诞生到今天的大规模使用,一路上有无数伟大的科学家为它付出了巨大的努力。
1913年,锂(Li)金属被发现具有最低的电极电位(−3.04 V)。然后,研究人员提出了层状氧化物正极材料二硫化钛(TiS2)与钴酸锂(LCO,简称LiCoO2)发生插层反应的新概念,实现了锂离子(Li+)的可逆插层/脱插层。直到用石墨作为负极材料,取代危险的锂负极后,锂离子电池(LIBs)作为历史上最伟大的发明之一,在1991年实现了商业化,并且以高能量密度、长寿命、可靠的安全性,深刻地重塑了我们的生活方式。近年来,便携式电子产品、电动汽车(EVs)和大规模储能的不断发展,推动了锂离子电池朝着更高能量密度、更快充电速度、更低的成本方向发展。
特别是在电动汽车领域,尽管近年来电动汽车在各种技术和低成本方面都取得了良好的发展,但仍缺乏消费者的接受度。因此,目前市场对电动汽车的接受度相对较低。其中一个关键原因是电动汽车中LIBs的快速充电能力不足,因此电动汽车的充电时间比内燃机汽车(ICE)要长得多。因此,提高锂电池的快充能力将是满足社会需求、推动电动汽车走向市场的强大推力。
国际上诸多组织和机构都提出了实现快速充电的目标,如美国能源部提出的极限快速充电(XFC),旨在促进纯电动汽车(BEVs)的充电体验可以与ICE汽车3-5分钟的充电体验相媲美。此外,美国先进电池联盟(USABC)的目标是用于电动汽车应用的高性能电池在15分钟内获得80%的荷电状态(SOC),可用系统能量为45千瓦时,还可以进行1000次可用能量动态压力测试。
然而,要实现快速充电的目标仍有一定难度。许多研究指出,限制快充容量的关键在于电池本身,尤其是电池的负极。为了节省时间,电池在高速充电时不可避免地会承受更大的电流。虽然节省了充电时间,但电池会受到更显著的影响。具体来说,高倍率充电会引发电析锂、机械效应、发热等一系列副反应,是加速电池老化的罪魁祸首,甚至造成电池不可控制的安全问题。
在LIBs的应用方面,LIBs在高纬度/高海拔寒冷地区的应用和特殊应用(如极地/航天探险和军事领域)仍然具有挑战性。在这些领域或应用中,电池需要在−20°C,甚至−60°C以下工作。但LIBs的最佳工作温度应控制在15 ~ 35℃。低温严重限制了充/放电倍率,这是由于传质和界面电荷传递过程的动力学缓慢所致。此外,与正常的插层行为相比,低温下不良动力学过程产生的较大极化更有利于石墨上的析锂。
电能和热量的滥用是电池热失控的关键原因。电池的安全性能和热稳定性会因为电能的滥用而降低。而且锂金属与电解液之间的副反应是电池过充电时热稳定性下降的主要原因。同时,隔膜会被锂枝晶刺穿,造成局部短路,从而导致电池热失控。鉴于LIBs中析锂对电池的安全性能和快速充电能力的起着决定性的作用,因此研究锂离子锂的析锂机理并进行及时监测具有十分重要的意义。
具体地说,“析锂”一词通常是指金属锂在石墨负极上的沉积。Li在石墨中形成GICs(石墨插层化合物的简称)的第一阶段LiC6的热力学电势与Li金属的热力学电势非常接近(0 V vs. Li/Li+),导致锂金属很容易沉积在石墨颗粒上,特别是在高充电倍率、高荷电状态、低温和滥用的情况下(见图1)。此外,枝晶状锂会导致活性锂的损失并诱发电池内部短路,增加电池的电阻,甚至造成安全问题。
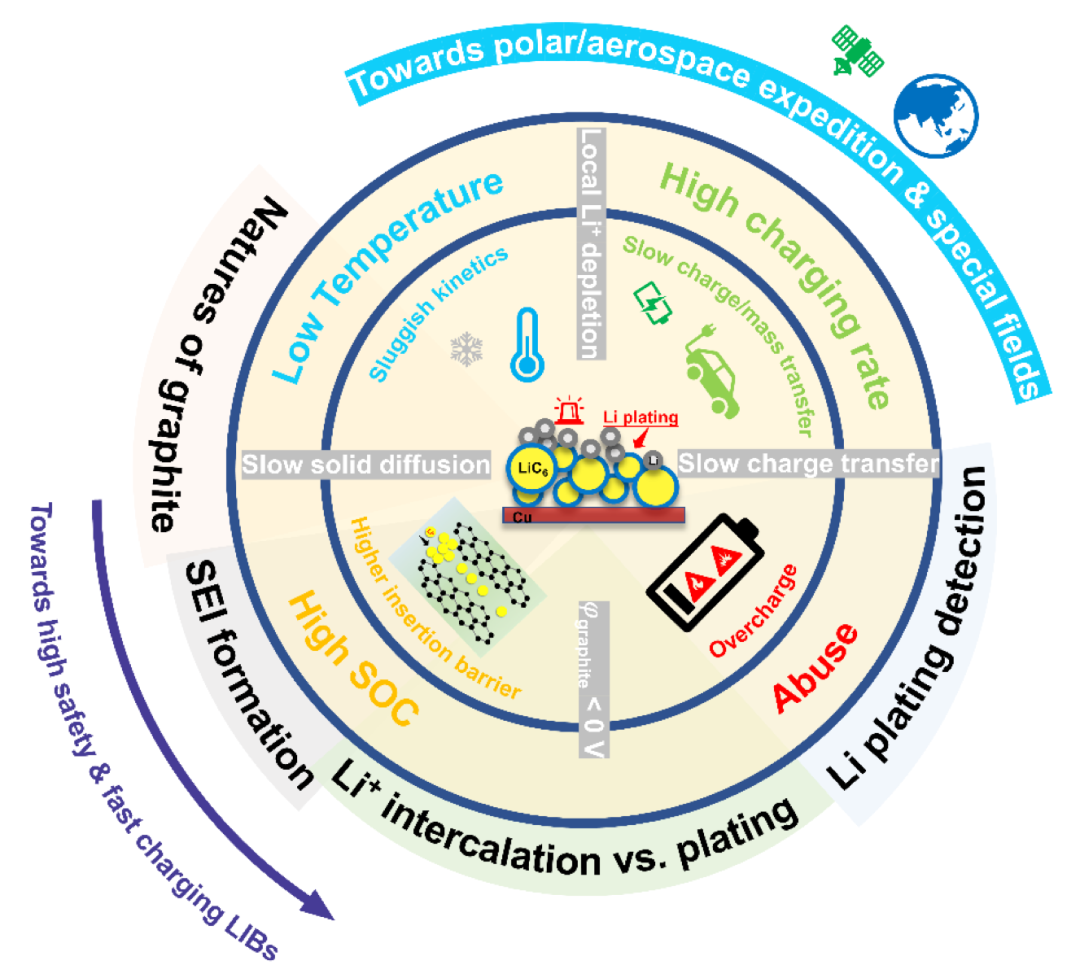
图1 石墨负极析锂诱因
目前关于析锂的研究已经有很多,但关于析锂的研究历程以及与石墨结构和界面化学相关联的分析报道很少。本文介绍了石墨的结构和界面化学,梳理了国内外学者对锂插/镀机理的认识。本文首先介绍了有利于锂可逆插层/脱插层的石墨的结构性质。其次,本文分别综述了有电解液和无电解液时锂嵌入石墨的历程。需要注意的是,在第一循环充放电过程中会形成电极/电解质界面相(EEI),这对Li+的插入和析出有很大的影响。因此,研究SEI的形成、组成和结构至关重要。在SEI形成过程中,Li+插入石墨也在发生。本文也详细介绍了锂离子嵌入石墨的过程,并讨论了限制锂离子嵌入石墨的因素。此外,本文综述了插锂动力学和析锂的研究过程,并讨论了影响析锂的4种重要机理,并进一步分析了锂离子在极端条件下(高充电倍率、低温、高荷电状态)的锂离子沉积机理。本文还总结了析锂造成的不良影响和析锂的检测方法,以期预测锂沉积的发生。最后,对LIBs中析锂的后续研究方向进行了展望。
我们对石墨有什么了解?
众所周知,石墨负极的工作电位(~0.1 V vs. Li/Li+)在热力学方面高于0 V (vs. Li/Li+)。石墨的理论比容量高达372 mAh g−1,因此石墨仍然是迄今为止最常用的LIBs商业化负极材料。如图2a所示,石墨具有典型的层状结构,每一层都由碳原子sp2杂化形成共价键,使每个碳原子可以在同一平面上以120°角与相邻的三个碳原子相连。同时,化学键是交联和延伸的,因此,六个碳原子形成一个规则的六边形碳环。碳环在平面上扩张形成石墨片层结构。与此同时,每个碳原子剩余的未杂化p轨道与电子相互作用形成π键,允许电子在石墨层之间自由移动,因此,石墨具有良好的导电性。由于π电子能吸收可见光,所以石墨会呈黑色,而且相同碳层之间的碳原子是共价键连接,所以很难破坏它们。但在垂直方向上,两层之间的距离为3.354 Å,是平面层中两个碳原子之间的距离的两倍多。因为相邻的两个碳层在弱范德华力的作用下结合。这种结构可以允许外来化学物质(如某些原子、分子、离子或复合离子等)在不破坏碳层中的石墨网络结构的情况下顺利嵌入,形成GICs(石墨插层化合物的简称)。那些插入石墨层之间的物质被称为“客体”,石墨充当“宿主”。客体嵌入石墨主体的反应称为嵌层反应。虽然在2H和3R石墨相中存在两种不同的层序,但两种石墨相的插层机理和容量是相似的。
本文重点研究了锂离子与石墨之间的的相互作用,特别是锂离子在石墨层中的插入状态。随着锂的插入,石墨经历了一系列相变。不同的相,也称为阶段,根据锂在石墨中的浓度来定义。假设Li的插层没有面内顺序,如果LixC6中x(Li)<0.25,则经历1L、4L、3L、2L阶段的稀释区。锂化程度较高的相,如阶段2和阶段1(分别为LiC12和LiC6)被分配到空间群P6/mmm,对应于石墨烯层的堆叠从AB序列转移到AA序列。不同的Li插层阶段和相变过程如图2b所示。许多研究表明,每个阶段(包括1L、4L、3L、2L、2和1阶段)的数字是指两个已占用层之间的未占用石墨烯层数。阶段1L, 3L, 2,1的颜色和结构如图2c所示。通过X射线衍射(XRD)和电化学测试,C6完全锂化为LiC6时,总体积膨胀率为13.2%另一个有趣的发现是,从C到2L阶段,层间距dC-C的增加具有准线性行为,但在2L到2阶段,层间距几乎没有变化。在阶段1中,dC-C显著增加,这是由于每个石墨烯层中都嵌入了Li。当Li与石墨形成LiC6时,相邻石墨烯层间的层间距离增加了~ 10%(见图2d)。
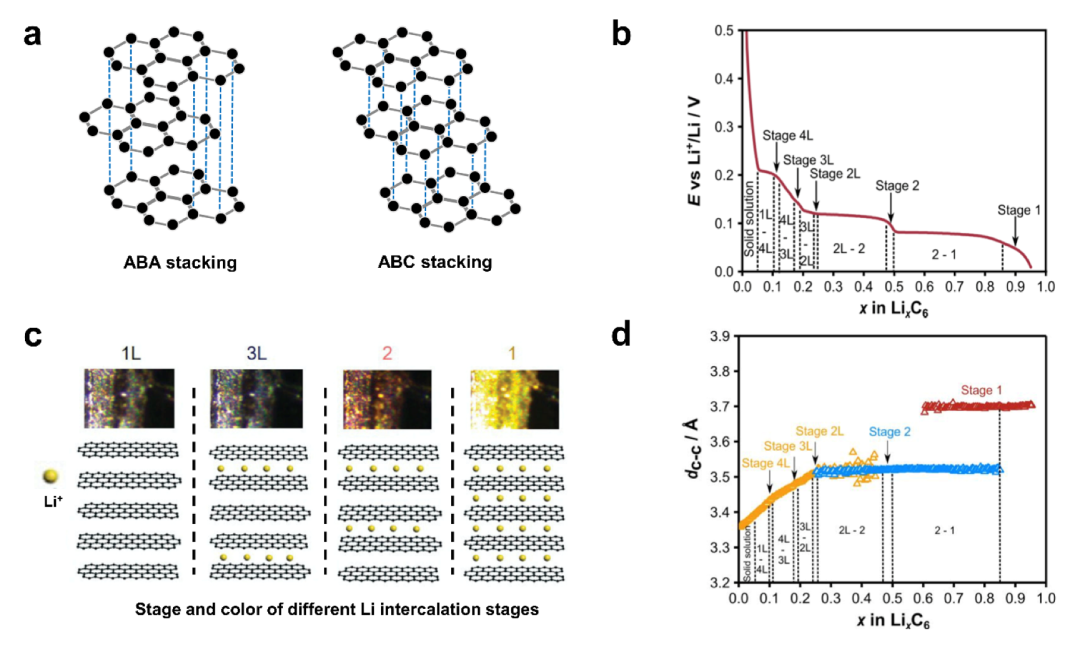
图2 a) ABAB (2H)和ABCABC (3R)两种不同堆叠顺序的石墨结构示意图。而ABA堆垛是石墨的主要类型。b)第二次循环中石墨不同的锂插层阶段和相变状态。c) 1L、3L、2,1阶嵌锂石墨的颜色和结构。d)第二次循环后石墨层间距dC−C在不同锂含量时的分布。
我们对锂嵌入石墨的过程有什么了解?
锂与石墨的相互作用
一些研究人员利用Surf. Sci.方法研究了吸附锂在石墨单晶表面的扩散科学。研究发现,被吸附的Li原子在石墨表面是可移动的,即使温度低至−173℃。此外,据报道,由于锂的亚单分子层是无序的,因此锂枝晶易于生长。Jäckle和Groβ更进一步,通过ab-initio计算研究了Li(001)表面上吸附的Li原子,模拟了几个Li原子层已经沉积在石墨表面上的情况。并将Li的枝晶生长归因于相邻吸附Li原子间较弱的相互作用和Li在Li(001)表面的扩散特性,这有利于Li在石墨表面和金属Li表面的非光滑生长。此外,在超高真空条件下利用Li原子蒸发到HOPG表面,研究了Li在HOPG单晶中的化学插层过程。通过奥格尔电子能谱(AES)和表面依赖的化学反应,报道了亚单层覆盖的Li扩散到石墨中,其活化势垒仅为0.16±0.02 eV。最近,有学者通过原位拉曼光谱测量研究了锂离子在石墨中的层呼吸模式,得到了锂离子在石墨中嵌入/脱嵌入的详细经验证据。通过使用原位测量,通过明显的低频拉曼特征观察到LiC18, LiC12和LiC6相,这是由诱导局部应变引起的石墨晶格位移引起的。此外,在深荷电条件下,由于插层Li施加应变,锂化石墨具有石墨烯样特征。
虽然上述研究仅考虑了锂与石墨之间的反应,而没有电解液的存在,但也为研究锂离子插入石墨的动力学过程提供了思路。此外,据报道,由于石墨颗粒没有足够的时间达到平衡,Li+嵌入石墨的过程以收缩核的形式进行。
值得注意的是,锂离子在插层过程更倾向于发生在石墨的端面,而不是在基面,因为脱溶剂和Li+进入石墨晶格的能垒在边缘面为0.3~0.7 eV,而基面为~ 10 eV。虽然Yao等人报道了不同缺陷下通过基面的能垒可以降低到6.35-2.36 eV,但与通过端面扩散的能垒相比,仍高出一个数量级。因此,重要的是要,与基平面相比,Li+更有可能通过边缘平面嵌入石墨。Gao等人利用原位光学显微镜研究了单个石墨颗粒的Li插入过程,发现Li插入到石墨的相变是以收缩核的方式进行的。另一个令人惊讶的发现是石墨颗粒的边缘面先变色,然后颜色向内移动,这与Li+更容易通过石墨的端面扩散是一致的。同时,该研究还观察到金属锂首先沉积在石墨的端面上。
石墨负极充电过程
SEI:形成,组成和结构
在LIBs的充电阶段,Li+从正极脱出,通过电解液和隔膜到达负极。同时,正极电位升高,负极电位下降,电池的整体电压升高。在电池放电过程中,Li+从负极返回到正极,降低了电池的电压。随着电池内电势的增大/减小,电解液与电极之间也发生电化学反应,固相氧化/还原产物堆积会形成界面膜。界面膜的稳定性对电池的循环性能至关重要。一方面,在界面膜形成的过程中,会消耗掉一部分Li+,导致电池容量的损失。另一方面,界面膜具有离子传导和电子绝缘的特性,可以保证Li+的快速迁移,防止电解液组分的持续分解,也可以防止溶剂分子共插层对电极材料造成不可逆破坏。
需要指出的是,界面膜可以统一称为电极/电解质界面相(EEI),在负极表面形成的界面膜通常被称为固体电解质界面相(SEI)。对于石墨负极,随着表面电位的降低,各种成分(阳离子、溶剂、阴离子、电解质添加剂、微量水)在电极表面竞争性反应,并还原生成SEI。还原反应的顺序和速率取决于各组分的还原势、浓度、反应活化能等因素。An等人介绍石墨表面SEI形成电位范围为0.8 ~ 0.2 V(vs. Li/Li+),还原反应产生的有机/无机不溶产物沉积在石墨负极表面,可溶性产物扩散到电解质中(图3a)。如果SEI没有完全形成,并且石墨负极电位持续下降到0.2 V以下(vs Li/Li+),则溶剂分子和Li+周围的溶剂化基团将共同插入石墨层之间。如果共插层过多,石墨层就会剥落,对石墨负极的稳定性造成不可逆的损失。
Liu等使用电化学石英晶体微天平(EQCM)、差分电化学质谱仪(DEMS)和原子力显微镜(AFM)原位监测了SEI在HOPG表面的形成过程。如图3b和3c所示,作者采用循环伏安法(CV)从开路电位(3.0 V vs. Li/Li+)扫描至0.0 V(扫描速率1.0 mV s−1),将SEI的形成过程分为四个阶段。
(1)当电压从2.0 V下降到0.91 V时,在这一阶段石墨表面可以发现不规则的岛状沉积,对应于LiF的形成,表明LiF可以通过化学或电化学的方式发生。
(2)在0.91 V ~ 0.74 V阶段,溶剂分子和Li+形成的溶剂化结构共插到石墨层中。
(3)0.74 V ~ 0 V阶段,与石墨共插层的初始溶剂化鞘中的碳酸乙烯酯(EC)开始减少,沉积在石墨上的颗粒开始大量增加并发生聚集,这可以认为是以LEDC为主的SEI开始形成。
(4)在随后的正向扫描阶段,当电压上升到0.3V以上时,SEI中的部分有机组分发生再氧化。
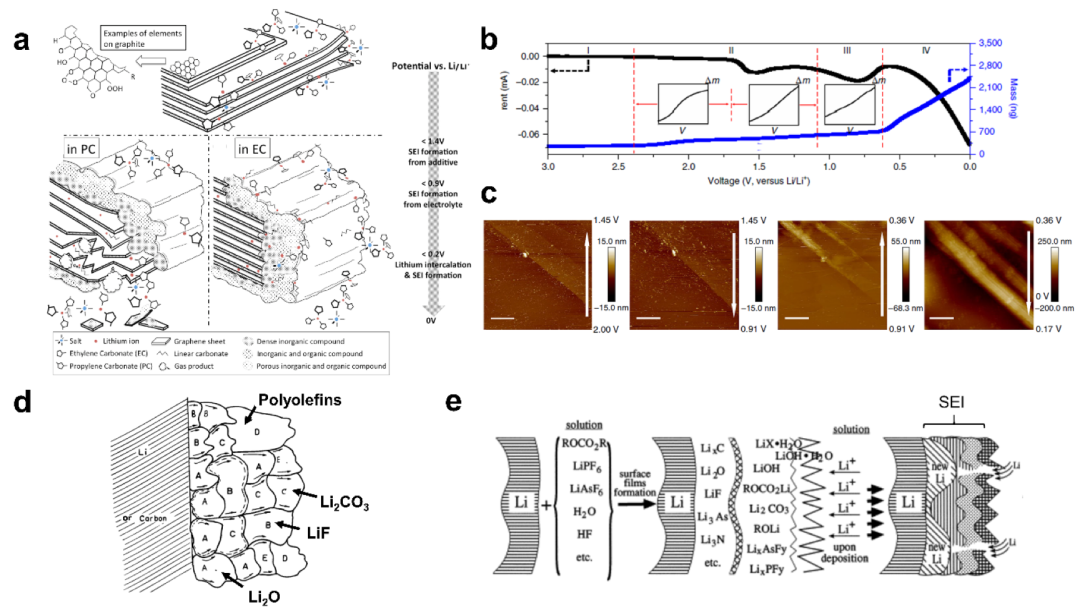
图3 a)石墨表面SEI形成过程示意图。b)石墨表面SEI形成过程中电位和质量变化曲线。c)电位变化时石墨负极表面的原位AFM图像。d)用传统的马赛克模型描述具有多种有机/无机组分的SEI。e) SEI层状结构模型示意图。
许多学者对SEI的主要组成部分进行了初步的研究,并对简单的LiF和LEDC进行了验证。然而,由于初始SEI在反复循环过程中可以演化成极其复杂的化合物/混合物,因此初始SEI中的产物极其复杂。这种复杂性的产生主要有两个原因。第一个原因是初始SEI的化学成分和结构不稳定。其中,被认为是EC还原后SEI的初始组分LEDC可被再氧化。值得注意的是,应该指出的是,LEMC,而不是LEDC,后来被证明是SEI的主要成分,因为LEMC与离子绝缘的LEDC相比具有更高的离子电导率。其次,SEI对周围的各种物质具有化学敏感性。例如,从正极溶解出的过渡金属离子,溶剂氧化产生的二氧化碳等等,这些可以改变SEI的结构和组成,从而导致SEI中复杂的化合物,如氟磷酸锂、羧酸锂、聚乙烯氧化物、Li2O、LiF等。
关于SEI的结构模型方面,人们早在1970年开始关注负极表面膜的结构和组成。1977年,Dey等人试图对锂表面形成的物质进行表征。从1990年开始,SEI的概念从锂金属负极扩展到石墨负极。1997年,Peled等提出SEI的马赛克模型。随后,Aurbach等人在1999年提出了SEI的层状模型。如今,马赛克模型和层状模型是描述SEI的两种主要模型。
在描述SEI的马赛克模型中,电解液组分还原产生的有机和无机产物随机分布在电极表面,如图3d所示。然而,非均质镶嵌结构的形成会影响Li+的传输路径,因为Li+总是沿着迁移速率较高的通道迁移。由于SEI丰富组分的迁移速率不同,难以实现Li+的均质插层/脱插层过程,从而导致枝晶Li和死Li。SEI的层状模型中,电解液组分的还原产物均匀分布在电极表面,形成层状结构,如图3e所示。一般由低氧化态的无机内层(如LiF、Li2O Li2CO3、LiOH等)和高氧化态的有机外层(如ROCO2Li、ROLi等)组成。在SEI的初始形成阶段,电解液组分首先被还原为有机产物,较靠近电极表面的组分随后被还原为相对稳定的无机物,这些无机产物极大地影响了SEI的电子绝缘性能。
在分析仪器的帮助下,我们可以通过信号的反馈来监测EEI的演化过程。但要探讨EEI的形成机制,还需要从更微观的角度进行解释。最近的研究发现EDL(电双层)的初始结构对EEI形成有较大的影响,即内亥姆霍兹层对Li+的特性吸附和Li+的溶剂化行为会共同影响EEI的形成。电极表面存在的初始吸附决定了EEI的初始化学成分和结构,而Li+的溶剂化结构参与了EEI的动态演化,它们之间的相互作用共同影响了EEI的稳定性。
一般认为,如果电极与另一电极相接触,金属或氧化物就有失去电子直至达到动态平衡的趋势。电极与溶液中过量电荷积聚的界面面积定义为EDL。EDL的概念由von Helmholtz于1853年提出并建模,Gouy、Chapman和Stern对其进行了进一步优化。Stern将EDL分为紧密层和扩散层。致密层包括内亥姆霍兹层(IHP)和外亥姆霍兹层(OHP)。在电极的初始状态下,IHP中存在对溶剂分子和阴离子的特异性吸附。受到IHP中有限空间的限制,其中不能包含溶剂化基团。而OHP中没有特异的吸附现象,但存在具有溶剂化结构的基团。亥姆霍兹平面是特异性吸附离子、溶剂化结构等反应底物离电极最近的位置,也是初始溶液中离子和电子氧化/还原反应的主要场所。不可否认,大多数阳离子(Li+, K+, Na+, Al3+等)具有较强的与溶剂分子络合形成溶剂化基团的能力,而阴离子则由于与溶剂分子的络合能力差而难以被溶剂化。因此,可以得出阴离子在不与溶剂结合的情况下更有利于吸附在电极表面。在目前的研究中,深入了解电极表面内亥姆霍兹层的特征吸附对形成稳定界面起着重要作用。
Yan等报道EDL中的特异性吸附可能只存在于电池的第一个循环中,之后它将被电子绝缘的EEI所取代,成为电极与电解液相之间的重要桥梁。电极IHP离子特异性吸附的初始状态直接关系到电极/电解质界面的氧化还原反应,从而影响EEI的结构和化学成分。
除了特异性吸附外,Li+的溶剂化结构是影响EEI的另一个重要因素。对于EEI的形成过程,电解液中的溶剂化结构相对EEI的形成有显著影响。最近的证据表明,在低浓度电解质中,Li+倾向于与强极性溶剂分子形成溶剂化结构,而阴离子则被排除在溶剂化鞘中。然而,在高浓度电解质中,阴离子更容易进入溶剂化鞘。与链状溶剂分子和阴离子相比,环状溶剂分子在稀溶液中与阳离子(Li+、K+、Na+、Al3+等)配位时更容易形成溶剂化结构。而这些参与溶剂化结构的电解质组分,特别是进入内溶剂化层的电解质组分,会参与EEI的形成,从而影响EEI的结构、组成、离子电导率和厚度。Yao等成功制备了弱溶剂化电解质(WSE),该电解质在低盐浓度下实现了电解液中存在大量离子对和聚集体,并在低盐浓度下使更多阴离子参与到溶剂化鞘中。并在石墨电极上形成独特的阴离子衍生电极界面,可实现快速充电和长期稳定循环的性能。通过第一性原理计算,作者也揭示了阴离子和溶剂分子在溶剂化结构中对Li+的竞争配位,为今后新型电解液的开发和设计提供了新的思路。
Li+嵌入石墨
在充电过程中,Li+会从正极脱出,然后穿过电解液插入到石墨负极中。但是,如上所述,在Li+插入石墨之前,会在石墨电极上形成SEI,这对Li的插入和析出有很大的影响。
众所周知,石墨负极的充电过程在原子尺度上可以分为4个连续的步骤(图4a):
(1)溶剂化的Li+在电解液中移动,特别是通过石墨电极中的曲折通道和微孔。
(2)溶剂化的Li+一旦到达石墨/电解质界面,由于存在电子绝缘SEI,电荷转移不能立即进行。Li+的溶剂化鞘层在穿过SEI之前必须被剥离,这被称为解溶剂化过程。
(3)裸露的Li+通过迷宫般的SEI进行扩散并嵌入石墨层。
(4)Li+在石墨内扩散,伴随电子转移和石墨晶格重排(由AB层叠向AA层叠)。
根据上述4个步骤,将Li+更快地插入石墨的限制因素大致可分为以下两类:
(a)质量输运:主要包括Li+在电解质和碳材料中的扩散,本例为(1)和(4)。
(b)电荷转移:Li+在SEI上的迁移可以形成一个主要的动力学势垒,对于石墨负极,该势垒由(2)和(3)组成。因此,解耦质量输运和电荷转移的影响来确定速率决定步骤是必要的。
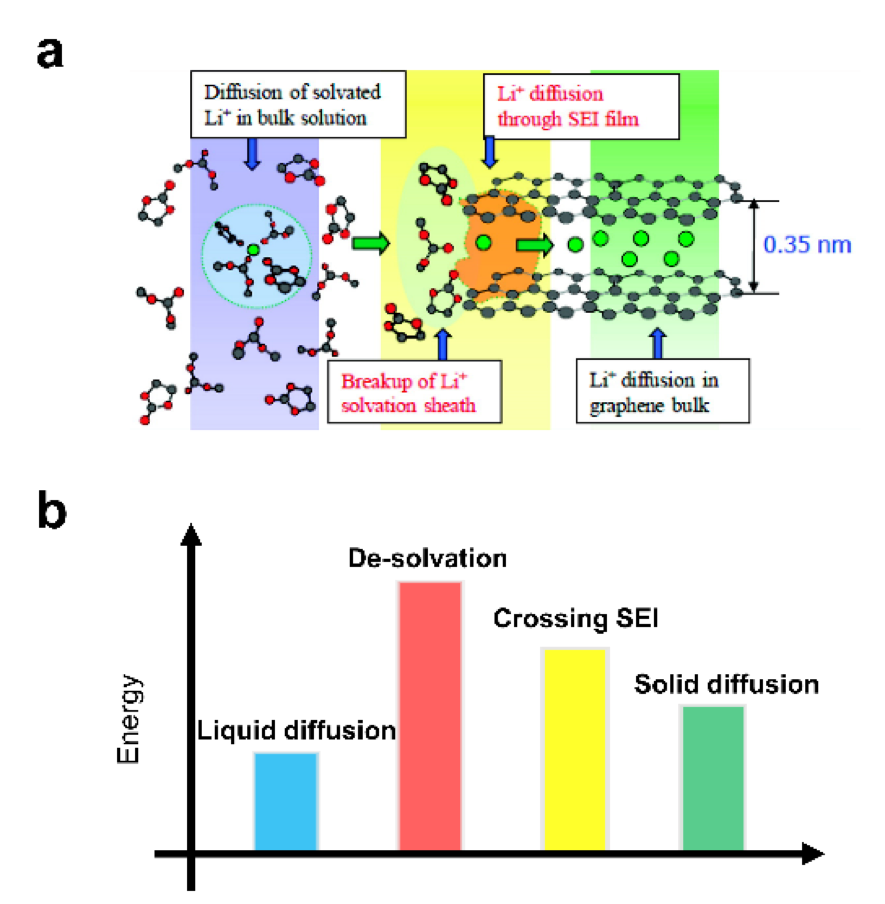
图4 a)充电时锂离子嵌入石墨过程示意图。b)对应于(a)连续4步的能垒图。
关于两个限制因素的能垒(图4b), Li+在石墨晶格中的固体扩散受到20-40 kJ mol−1范围内的能垒限制,据报道,该能垒随着锂化程度的增加而增加。然而,当使用碳酸盐电解质时,各种正极材料和石墨的脱溶剂能相似(50-70 kJ mol−1)。但脱溶剂化的能垒可随电解液的组成和温度而改变。然而,到目前为止,关于Li+穿过SEI的能量势垒的讨论很少,因为SEI的性质随着电解质的组成而发生了巨大的变化,特别是对于溶剂、锂盐和电解质添加剂。例如,曾经有报道称Li+穿过SEI的能垒约为20 kJ mol−1,但没有更可靠的研究和后续的研究继续证实这一点。
到目前为止,对于质量传输和电荷转移是充电过程中的主要限制因素,人们的看法还不太一致。首先,电池测试中有很多变量(如充电倍率、工作温度、电极材料和厚度等),可以改变主导因素。对于较厚的电极或较大电流密度的电极,质量输运在整个插层动力学中起主导作用,而对于较薄的电极,特别是在零下温度下,由于活化能的增加,电荷转移可能成为限制因素。因此,在区分限速步骤时,需要强调电池测试条件。其次,脱溶剂化过程不能被认为是反应速率的决定步骤,而它是最需要活化能的步骤。根据阿伦尼乌斯方程,这是因为速率常数不仅是活化能的函数,而且还包括指前因子。也就是说,除了能垒之外,速率常数是决定化学反应速率的另一个指标。
在此基础上,我们可以总结为:(1)锂离子嵌入石墨的过程与各种电池参数(电极厚度、电极材料、温度、面积负载等)有关。在宣布充电速率决定步骤之前必须给出测试条件。(2)如果两个过程贡献相似,则不存在速率决定步骤,可以通过多个步骤同时控制充电过程。(3)应重视进一步的机理研究和探索速率决定步骤的实验设备。从理论上或实验上寻找决定速率步长的关键因素,都将提供更加可靠的结论。
(未完待续)